信息中心



2024年9月19日,沐鸣开户劉智攀教授團隊在Journal of American Chemical Society (JACS)期刊上發表了一篇題為“In Situ Surfaced Mn–Mn Dimeric Sites Dictate CO Hydrogenation Activity and C2 Selectivity over MnRh Binary Catalysts ”的研究成果(https://doi.org/10.1021/jacs.4c10052)🪣。論文為課題組近期利用基於LASP軟件的人工智能原子模擬在Ag基乙烯環氧化(Nature Catal. 2024, 7, 536)🧘🏽♀️、稀土CeZrO氧化物催化(JACS,2024🧎🏻➡️◾️,146,10822)等復雜催化體系研究中取得的又一重要成果。
該成果報道了基於機器學習全局勢函數大規模原子模擬🛬,在CO氫化的實驗條件下(523 K🦝,20 bar,H2/CO = 2),探尋MnRh二元催化劑的結構、反應活性和碳二含氧化合物(乙醛和乙醇)選擇性的最新研究成果。發現在實驗條件下,與體相的MnO相比,Mn和Rh在表面形成二元合金在熱力學上更加穩定💹,且會在吸附的O或OH的誘導下在Rh(211)的臺階位點上形成Mn–Mn的二聚位點。該位點有效降低了CO解離和碳二產物產生的能壘,提高了碳二產物生成速率和選擇性。
論文通訊作者是劉智攀教授🔽,張科想為論文的第一作者。項目感謝國家自然科學基金(12188101🚴🏻♂️、22033003、91745201),中央高校基本科研業務費專項基金(20720220011),國家重點研究發展計劃項目(2018YFA0208600)🪔👩🏻🔬,騰訊科學探索獎等的資助。
大規模乙醇工業合成一直是人類夢想。Mn助劑能顯著提高Rh基催化劑催化的合成氣(CO/H2)到碳二含氧化合物的活性和選擇性👩🏿🏫,但對MnRh催化劑的結構和Mn助劑在該體系中的作用🙎♂️,目前仍無統一定論🔇👱🏿♀️。雖然大部分的實驗認為Mn在該反應體系中主要以低價氧化物的形式存在(尤其是MnO)👍🏼,但高價態的氧化物和0價的MnRh合金的存在不能被完全排除😍。對於Mn的作用,目前的主要說法有兩個🧚🏽♀️✊🏿:(1)Mn能穩定傾斜吸附的CO✡︎,進而促進CO的解離⛲️;(2)Mn的氧化物能促進Rh+的形成💪🏼,而Rh+位點則被視為C-C偶聯發生的活性位點。要結束其中的爭議,重點在於弄清Mn在該體系中的存在形式和在催化劑中的所處位置,並找到反應的活性位點。
在這項工作中,作者應用了劉智攀課題組(zpliu.fudan.edu.cn)開發的大規模機器學習原子模擬軟件LASP (www.lasphub.com)👨🏻🦼➡️,創建了RhMnCHO五元全局神經網絡勢函數,對Rh(111)面和Rh(211)上不同Mn表面濃度的結構進行高效全局搜索。根據14種含不同Mn比例的Rh(111)和Rh(211)的穩定結構🕖,建立了523 K下MnRh體系的熱力學相圖,確定了在典型的實驗條件下,Mn在表面與Rh形成二元合金比體相的MnO更加穩定(圖1)。隨後🫐,采用ML-TS大規模路徑搜索方法,遍歷了CO解離反應通道,發現在吸附的O或OH物種的存在下,次表層的Mn會被誘導到表面與O或OH成鍵🧑🏽🚒🧛🏽,而被誘導到(211)表面臺階位上的二聚的Mn-Mn位點能夠大大降低CO解離的反應能壘(圖2)。接著,以含有這種Mn-Mn的MnRh(211)作為重點研究對象,構建了CO氫化到碳二氧產物的全反應網絡(包含74種基元反應)🤌。作者發現和不摻Mn的純Rh(211)表面相比,二聚的Mn-Mn位點能穩定含不飽和O原子的物種的吸附,顯著降低CO解離和C2物種氫化的反應能壘,分別為0.77和0.59 eV,而對C-C偶聯反應影響較小🔌𓀂,能壘僅降低0.14 eV(圖3)👩🏼⚖️。通過電子結構的分析,作者認為這種二聚Mn位點之所以能夠穩定不飽和O原子的物種的吸附,是因為Mn在費米能級以上有大量的自旋向下的空軌道,促進Mn和O之間的反饋成鍵作用(圖4)。最後作者通過微觀動力學模擬🔪,表明二聚的Mn-Mn位點能大大提高CO氫化反應中碳二產物的活性和選擇性(圖5)。該研究結果在原子層面上揭示了MnRh催化劑在實驗條件下的結構和活性位點,對後續的合成氣製C2產物研究和催化劑設計具有指導意義。
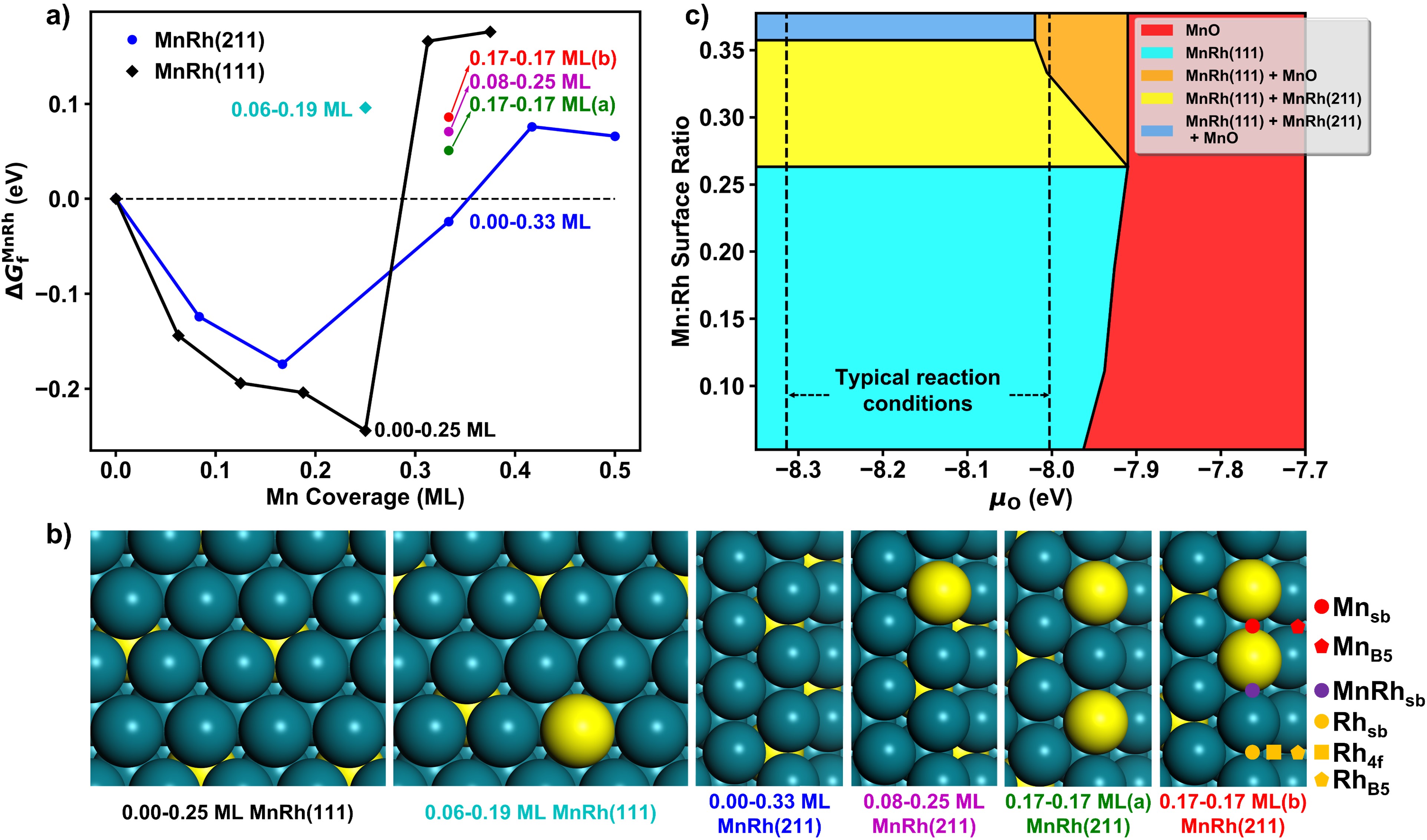
圖1:實驗條件下MnRh體系的表面熱力學相圖🌟。
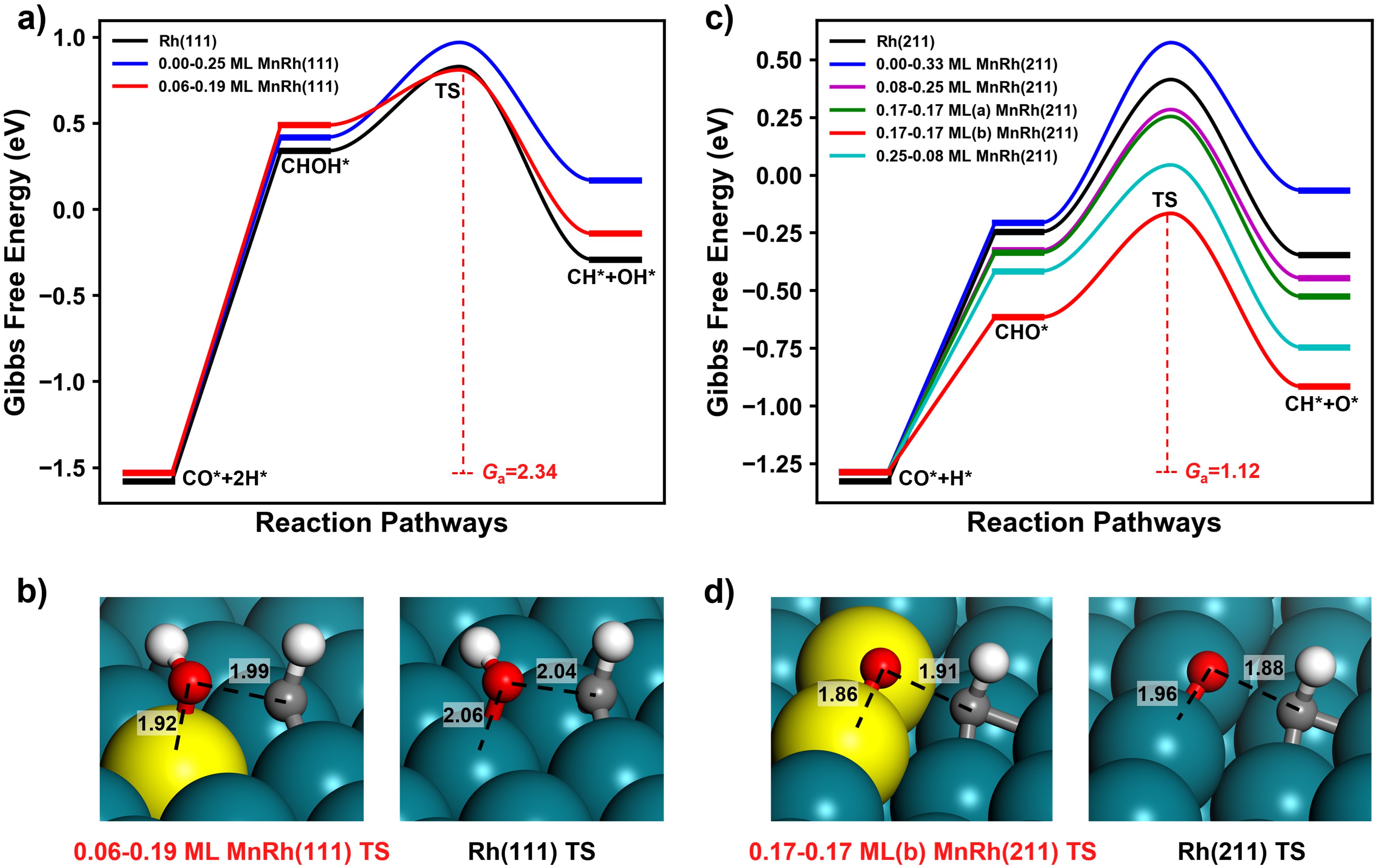
圖2👬🏻:Rh和MnRh表面的CO解離通道。

圖3:Rh(211)和MnRh(211)表面CO氫化反應具有最低能壘的反應路徑✍🏼。
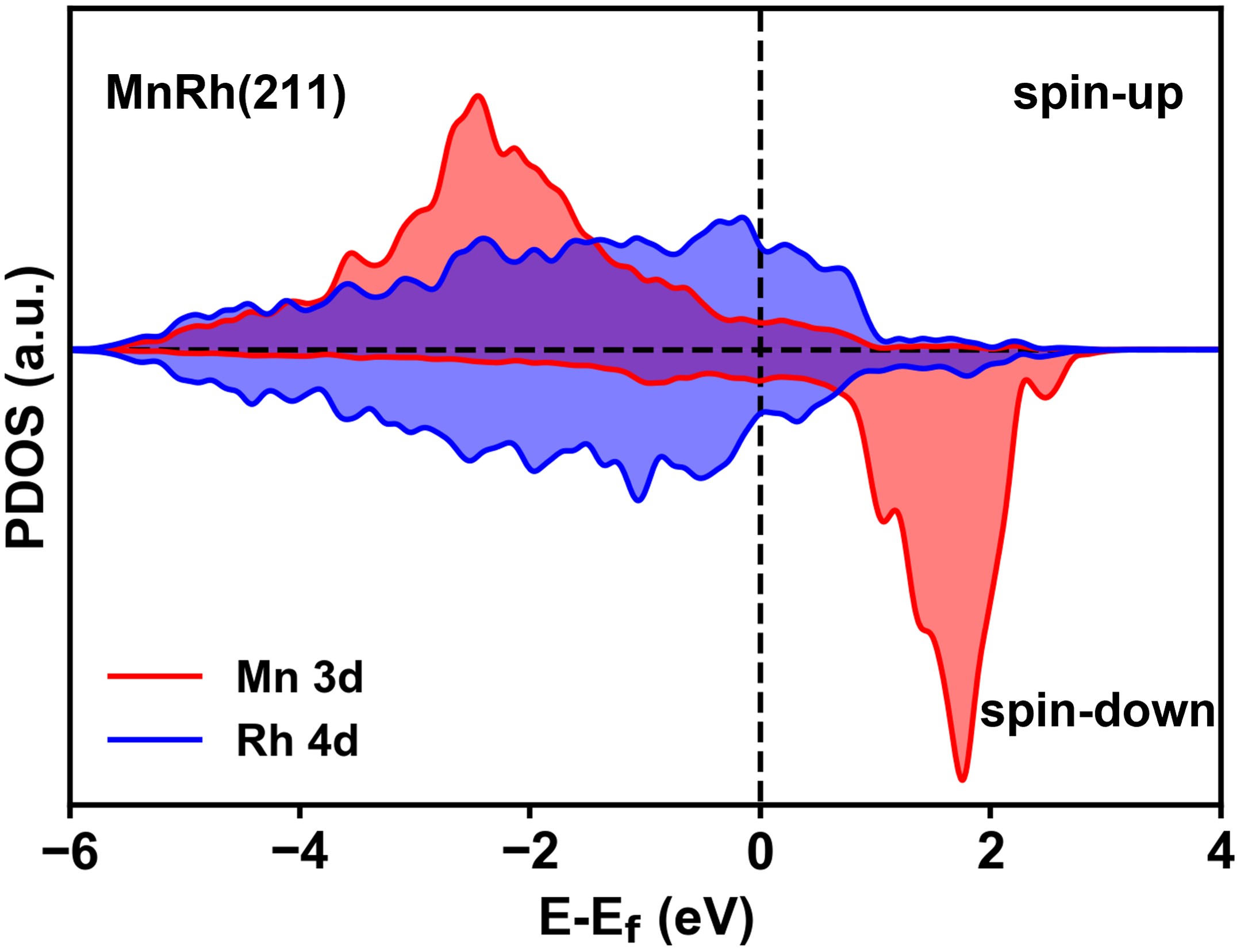
圖4:0.17-0.17 ML(b) MnRh(211)臺階位點上的金屬原子的投影態密度。
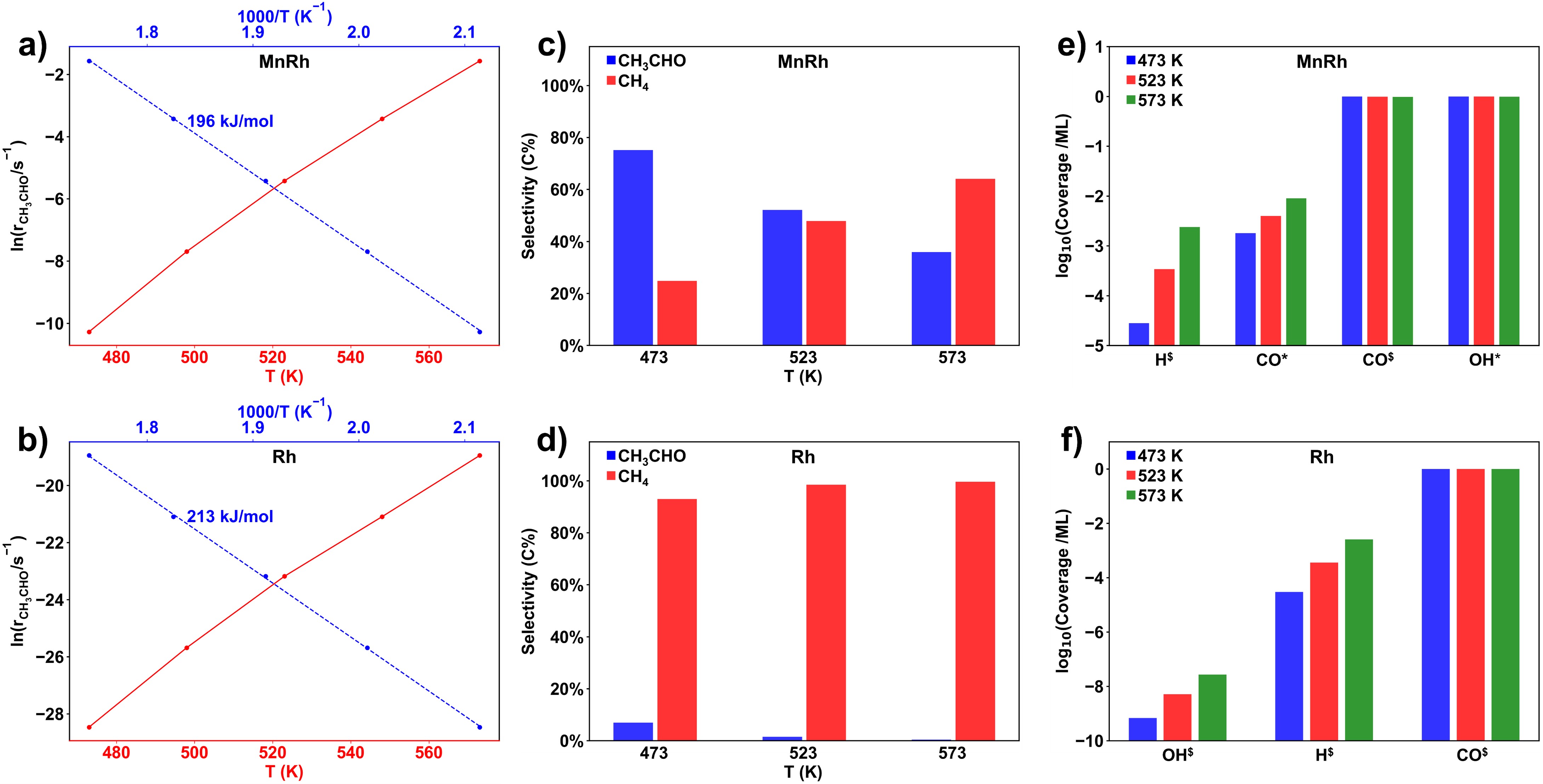
圖5:Rh(211)和MnRh(211)表面上反應的微觀動力學模擬🥇。


